Running GROMACS on HPC Systems
Environment
ITSO HPC4 Cluster
GROMACS versions: - Container-based: 2023.2 (NGC) - Source build: 2024.1
CUDA 12.4.0
GCC 13.2.0
OpenMPI
Apptainer/Singularity for containers
Issue
Need to run molecular dynamics simulations using GROMACS
Want to leverage GPU acceleration for faster simulations
Unsure which deployment method to choose:
NGC container (fastest for single node)
Source build (needed for multi-node jobs)
Resolution
SLURM Job Template
Create a SLURM job script with appropriate resource requests:
#SBATCH --job-name=gromacs
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#SBATCH --cpus-per-gpu=16
#SBATCH --gpus-per-node=l20:4
#SBATCH --partition=gpu-l20
#SBATCH --account=<account>
#SBATCH --time=01:00:00
#. Using NGC Container (Recommended for Single-Node Jobs)
Add these commands to your SLURM script:
export GMX_ENABLE_DIRECT_GPU_COMM=1
singularity run --nv \
-B <simulation_dir>:/host_pwd \
--pwd /host_pwd \
docker://nvcr.io/hpc/gromacs:2023.2 \
gmx mdrun -maxh 1.0 -ntmpi 8 -ntomp 8 \
-nb gpu -pme gpu -npme 1 -update gpu \
-bonded gpu -dlb no -nstlist 300 -pin on
Note
NGC container provides superior performance for single-node jobs
#. Building from Source with Spack (For Multi-Node Jobs)
Set up Spack environment:
spack env create gromacs
spack env activate gromacs
# Install GROMACS
spack add gromacs@2024.1%gcc@13.2.0 +mpi +cuda cuda_arch=89 ^cuda@12.4.0 ^openmpi
spack concretize -fU && spack install --only-concrete
Add these commands to your SLURM script:
spack env activate gromacs
gmx_mpi "<command>"
Performance Considerations
Hardware Performance Comparison:
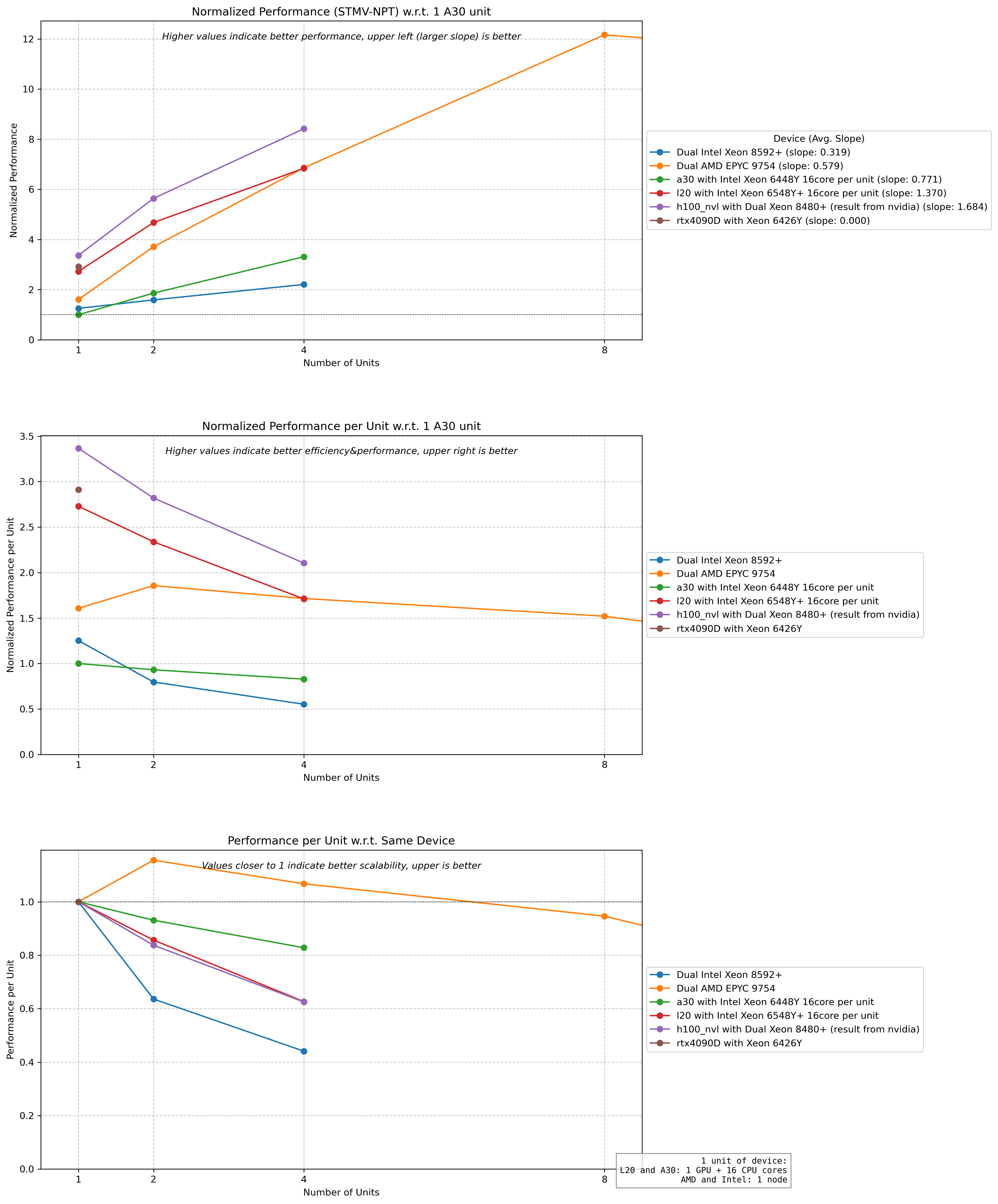
Combined Hardware-Threading Performance:
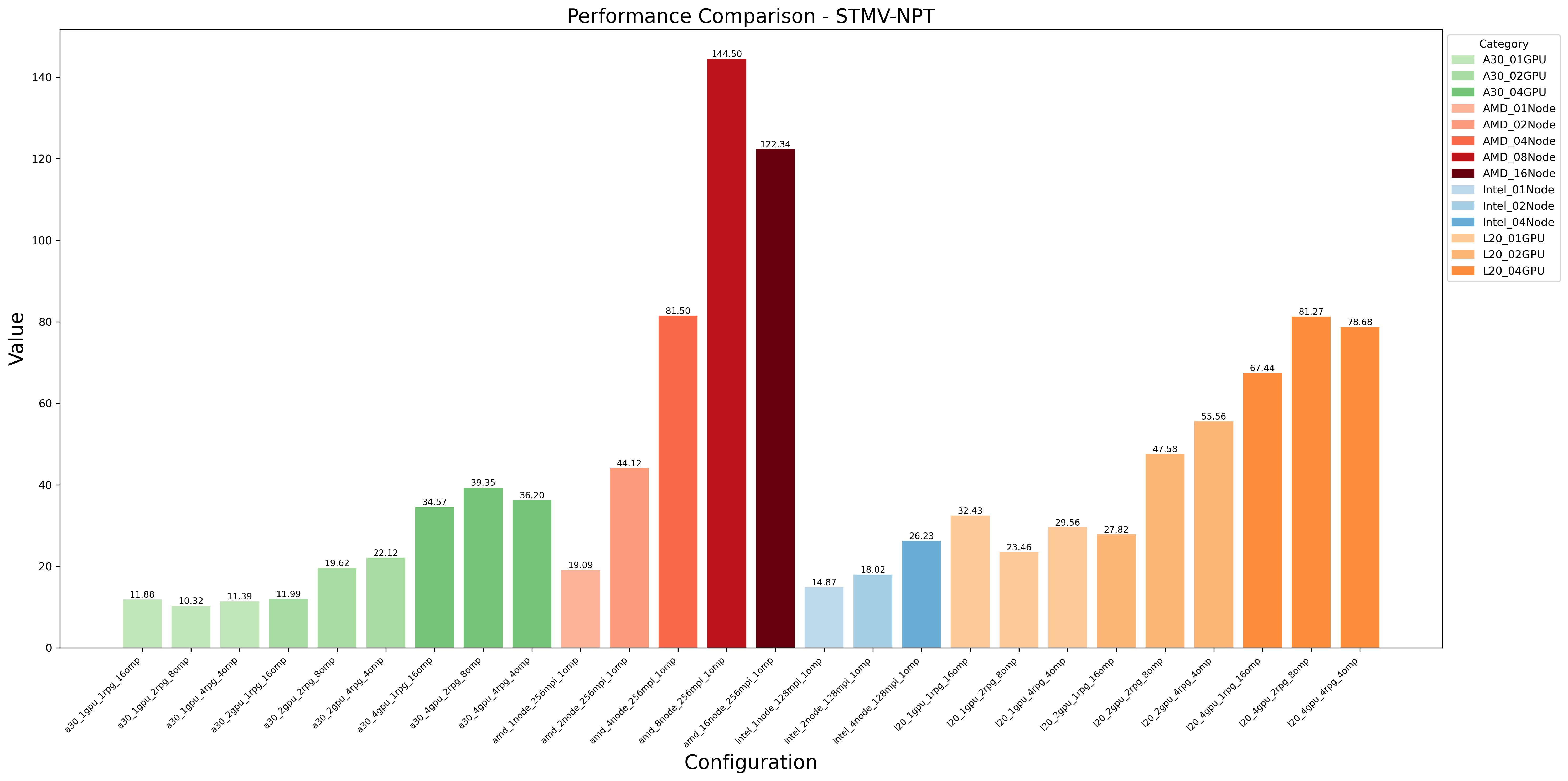
- Key findings:
One NVIDIA L20 GPU per job typically provides optimal cost-effectiveness
NGC container outperforms source builds for single-node jobs
Performance depends heavily on:
Number of MPI threads (ntmpi)
Number of OpenMP threads (ntomp)
Neighbor search frequency (nstlist)
Warning
Always benchmark your specific simulation setup to determine optimal resource allocation
Root Cause
GROMACS performance depends heavily on build configuration and runtime parameters. NGC containers are pre-optimized for single-node performance, while source builds provide flexibility needed for multi-node runs.
References
NVIDIA NGC GROMACS Container: https://catalog.ngc.nvidia.com/orgs/hpc/containers/gromacs
GROMACS Documentation: http://manual.gromacs.org/
Spack Documentation: https://spack.readthedocs.io/